Journal of Biomass to Biofuel (JBB)
ISSN: 2368-5964

Volume 1, Year 2014 - Pages 1-7
DOI: 10.11159/jbb.2014.001
Zeolite Catalyzed Hydrolysis in Aqueous and Ionic Liquid Media
Luisa M. Smallwood1, Robert L. White2
1Department of Chemistry, University of California-Berkeley,Berkeley, CA 94720-1460
2Department of Chemistry and Biochemistry, University of Oklahoma, Norman, OK 73019-0390
rlwhite@ou.edu
Abstract - Hydrolysis of ethyl acetate and starch in water and of cellulose in an ionic liquid by using HY and HZSM-5 solid acid catalysts are compared. In water, HZSM-5 is more active than HY. This trend is reversed in ionic liquid. Reactivity differences are explained based on interactions between solvent and catalyst acid site local environments, which depend on zeolite channel structure and Si/Al ratio. Greater hydrolysis reactivity is achieved when the ionic strength of the reaction media is high. This is a consequence of ion exchange, allowing for transfer of catalyst protons to the reaction media, where they can more effectively protonate ester and glycosidic functionalities. Water interacts more strongly with HY acid sites, presumably because the lower Si/Al ratio results in framework channels that are more hydrophilic.
Keywords: ethyl acetate hydrolysis, polysaccharide hydrolysis, ionic liquid media catalysis, liquid media zeolite catalysis.
© Copyright 2014 Authors - This is an Open Access article published under the Creative Commons Attribution License terms. Unrestricted use, distribution, and reproduction in any medium are permitted, provided the original work is properly cited.
Date Received: 2013-11-12
Date Accepted: 2013-12-18
Date Published: 2014-02-24
1. Introduction
Hydrolysis of biomass to release sugars that are subsequently converted to alcohols is one of several processes being investigated as a means to lessen dependence on non-renewable fuels. Biomass hydrolysis can be accomplished by using enzymes or through the use of acid catalysts. The use of solid rather than liquid acids is potentially advantageous because the acid could be removed by filtration and recycled after hydrolysis. Many solid acid catalysts have been evaluated for biomass hydrolysis under various reaction conditions [1]. Results from these studies suggest that catalyst effectiveness depends on numerous factors, making structure-activity comparisons and reaction mechanism elucidations difficult.
HZSM-5 and HY zeolites were among the first solid acid catalysts employed for hydrolysis in liquid media. HZSM-5 and HY are silica-alumina catalysts with well defined but different pore structures. Ethyl acetate hydrolysis catalyzed by these zeolites in 60 oC water revealed that the first order rate constant when HZSM-5 was employed was about 40 times greater than when HY was employed [2]. This large difference in catalytic activity was attributed to unspecified skeletal differences in the zeolite structures. Hydrolysis activity was found to vary with zeolite Si/Al ratio for both catalysts, with the optimum for HZSM-5 at about 47 and the optimum ratio for HY near 3. Dhepe, et al. [3] compared a variety of solid catalysts for hydrolysis by heating aqueous sucrose solutions for 6h at 80 oC and found that HZSM-5 had no activity (HY was not tested). They explained this finding based on steric considerations. The sucrose molecule was too large to enter HZSM-5 pores, making catalytic active sites inaccessible. In contrast, Jiang, et al. [4] reported that HZSM-5 was active for starch hydrolysis in water (22% conversion, 120 oC, 4h) and to a lesser extent for cellulose hydrolysis (<1% conversion, 150 oC, 40 min). Starch and cellulose molecules are much larger than sucrose, so these results appear to conflict with results reported by Dhepe et al. Interestingly, when cellulose was dissolved in an ionic liquid containing small amounts of water, HY exhibited a relatively high activity for hydrolysis (50% glucose, 130 oC, 2h) [5].
Results from studies of HZSM-5 and HY catalyzed hydrolysis under a variety of reaction conditions are presented here. Comparing the properties of these zeolites under identical reaction conditions provides insight into the importance of solid acid structure and solvent on catalytic activity. Detailed studies of reactions catalyzed by these zeolites lead to a better understanding of solid acid catalyzed biomass hydrolysis mechanisms and the roles of acid sites located within pores.
2. Experimental
Reagent grade chemicals and distilled water were used for hydrolysis reactions. Cellulose and 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl) ionic liquid were used as received from Sigma-Aldrich (St. Louis, MO). The sodium form Y zeolite (NaY) was obtained from Honeywell UOP (Y-54, Si/Al=5.30, Des Plaines, IL). NaY (1 g) was mixed with ammonium nitrate (0.25 mol) in water and the mixture was refluxed with constant stirring for 4 hours. The solid was filtered and then air dried prior to calcination at 550°C for 3 hours to produce HY. HZSM-5 (Si/Al=19) was synthesized by using the procedure described in the Mobil Oil U.S. Patent (US 3702886).
The mmol H+ available from the solid catalysts was determined by using a back titration method with phenolphthalein indicator. Catalysts (0.1 g) were added to a sodium hydroxide solution (0.01 M, 25 mL) and stirred for 10 minutes. Mixtures were then centrifuged and the liquid was removed. A 10mL aliquot of the liquid was then titrated with oxalic acid (0.01 M) to a pink to clear color change end point.
The ethyl acetate hydrolysis test solution was a mixture of 5% (vol) ethyl acetate (EA) and 5% (vol) propanol (P) in distilled water. Propanol did not react and was therefore used as an internal standard. The catalyst (0.01 g) and 5% EA/P solution (0.4 mL) were loaded into a quartz tube reactor and pressurized to a minimum of 2 MPa with nitrogen gas. The reactor was heated isothermally for 30 minutes at temperatures ranging from 70 to 150°C followed by immersion in a cold-water bath to quench the reaction. The reaction mixture was analyzed by using an HP 5890 gas chromatograph with flame ionization detection and a Carbowax fused silica column (Agilent 113-2132). All chromatographic analyses were performed with splitless injection.
For starch hydrolysis in aqueous media, catalyst (0.01 g) and 1% starch solution (0.4 mL) were mixed and loaded into the reactor and pressurized to at least 2 MPa with nitrogen gas and then heated isothermally at 150°C for up to 2 hours followed by immersion in a cold-water bath to quench the reaction. Samples for starch hydrolysis in Na+ enriched aqueous media were prepared in a similar manner except that the reaction medium was sodium chloride solution.
For cellulose hydrolysis in an ionic liquid medium, cellulose (0.1 g) was first dissolved in [EMIM]Cl (2 g) by using a temperature controlled oil bath to maintain a temperature of 120°C. After dissolution, the temperature was lowered to 105°C and distilled water (0.3 mL) was added in increments of 0.05 mL with constant stirring. Catalyst (0.25 g) was then added to this homogenous mixture. The hydrolysis reaction was carried out for 3 hours and the solution was sampled at 15 minute intervals for total reducing sugar assays conducted by using the Benedict's method [6].
Zeolite structures were determined by using a Rigaku Ultima IV X-ray diffractometer. Pattern matching and mineral assignments were made by using MDI JADE 9 software. Zeolites were also analyzed by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) by using a Mattson Instruments Nova Cygni 120 spectrophotometer (Madison, WI) and a modified Harrick Scientific Inc. (Ossining, NY) praying mantis diffuse reflection accessory with environmental chamber [7]. The DRIFTS sample temperature was maintained at 200°C under flowing He for 45 min to remove adsorbed water from catalyst surfaces. Infrared spectra were then measured at 200°C.
3. Results and Discussion
3.1. Ethyl Acetate Hydrolysis
Aqueous phase hydrolysis of ethyl acetate is commonly used as a test reaction for assessing the "water tolerance" of solid acid catalysts [8-13]. Typically, hydrolysis reactions are carried out with 5% ethyl acetate aqueous solutions at 60 or 70 oC for 2 hours. Ethyl acetate hydrolysis reactions were conducted with HZSM-5 and HY catalysts at temperatures varying from 70 to 150 oC in order to characterize their water tolerances over a wide temperature range. This temperature range was selected to extend into the range often employed for biomass hydrolysis. The molecular dimensions of ethyl acetate (ca. 4.95 Å) [14] are smaller than HZSM-5 and HY channels, so internal acid sites should be accessible. Hydrolysis was carried out for 30 min followed by analysis of the reaction mixture to measure the loss of ethyl acetate (i.e. conversion) and formation of ethanol and acetic acid products.
HZSM-5 exhibited increasing activity for ethyl acetate hydrolysis with increasing reaction temperature (Figure 1).
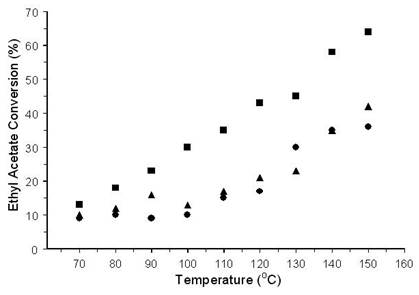
HY activity also increased with temperature, but was found to be comparable to a blank measurement (i.e. no catalyst). Catalytic activity associated with the blank measurement was due to an increase in H+ activity with increasing temperature, which is a consequence of larger water autoprotolysis equilibrium constants at higher temperatures [15]. The results shown in Figure 1 are consistent with reports by Kamiya et al. [13]and Okuhara et al. [16], who found that HY was inactive for ethyl acetate hydrolysis at 70 and 60°C respectively after 2 hours, and by Namba et al. [2], who reported that HZSM-5 performed better than HY for ethyl acetate hydrolysis in water. Drago, et al. [16] determined that all HZSM-5 acid sites are located inside zeolite channels. Therefore, ethyl acetate must diffuse into HZSM-5 channels and the ethanol and acetic acid products must diffuse out into the aqueous medium. Because HY supercage acid sites would be more accessible to ethyl acetate than those located in HZSM-5 channels, the greater ethyl acetate hydrolysis activity for HZSM-5 cannot be explained by steric effects. Concerning the lack of activity for HY, Kerr reported that HY can lose 25% of its original Bronsted acidity just by being exposed to water for 10-15 minutes [17]. In addition, Parker et al. [18] found that the HY framework could be destroyed by exposure to small amounts of water followed by heating to 300 oC. They postulated that water reacts to form AlOH and SiOH species at the expense of framework Al-O-Si bonds. When multiple Al-O-Si bonds are broken, extra-framework Al-O(OH) species may be formed [19]. This dealumination process destroys the HY framework. Although the HY catalyst used for the ethyl acetate hydrolysis experiments described here was not heated as high as 300 oC, which would have destroyed the zeolite channel structure, it is possible that some -Al(OH)n (n = 1,2) framework species were formed. Formation of -Al-OH and -Si-OH species would disrupt the zeolite framework in the vicinity of delocalized negative charges associated with acid sites, and could be responsible for the observed loss of HY acidity. This hypothesis is consistent with a report from Scherzer and Bass [20], who found that –Si-OH and –Al-OH groups formed by reactions with water did not interact with pyridine. In contrast to HY, HZSM-5 exhibited acid catalyst activity that was significantly greater than the blank. If water also reacted with the HZSM-5 framework, the effects were not as extensive as for HY. HZSM-5 may be more stable in water because of its larger Si/Al ratio, which makes it more hydrophobic than HY [21].
3.2. Starch Hydrolysis
Starch, a glucose polymer, is much larger than ethyl acetate, and should not pass into zeolite channels. Like ethyl acetate hydrolysis, HY catalyzed starch hydrolysis results were comparable to the blank (13 and 14% total reducing sugar yields respectively after 2 hours at 150 oC). However, HZSM-5 exhibited activity for starch hydrolysis that was significantly greater than the blank measurement (32% reducing sugar yield). Jiang et al. [4] also found that HZSM-5 was active for starch hydrolysis (120°C, 4 hr, microwave reactor), resulting in a 22% glucose yield. Considering that steric effects should prevent starch molecules from accessing HZSM-5 channels, this catalytic activity is surprising. The HZSM-5 acid site locations may play a role in its activity for starch hydrolysis. If HZSM-5 contains no external acid sites, as proposed by Drago et al. [16], starch molecules must have some access to internal sites. When a substrate is too large to enter HZSM-5 channels, the acid sites located near channel openings may still be accessible. Alternatively, HZSM-5 starch hydrolysis may result from reactions of H+ released from internal zeolite channels by ion exchange (vide infra).
3.3. Starch Hydrolysis in the Presence of Na+
Zeolite catalyzed starch hydrolysis occurs at much lower rates than ethyl acetate hydrolysis because starch molecules are too large to access internal acid sites. This limitation can be overcome by using ion exchange to release zeolite protons into the aqueous reaction medium [5]. A relatively high concentration of Na+ can be used to transfer H+ from zeolite inner surfaces to the aqueous medium, which causes a decrease in solution pH.
When zeolites were immersed in a 0.25 M Na+ solution, aqueous phase acidity was found to increase by ca. 0.3 mmol H+/g and 0.9 mmol H+/g for HZSM-5 and HY respectively, when compared to immersion in water. This is consistent with previous studies, which have shown that ion exchange with Na+ causes a decrease in the solid-state acidity of zeolites [22, 23]. The substantial increase in solution H+ concentration for HY may suggest that the presence of Na+ reverses the effects of water on framework bonds involving Al, but this is not confirmed by results presented here. The smaller increase in solution acidity for HZSM-5 may be explained by the difference in the Si/Al ratios for the zeolites. HY (Si/Al=5.3) had a much lower Si/Al ratio than HZSM-5 (Si/Al=19), so there were fewer Al atoms present in HZSM-5, and therefore fewer acid sites that could participate in ion exchange.
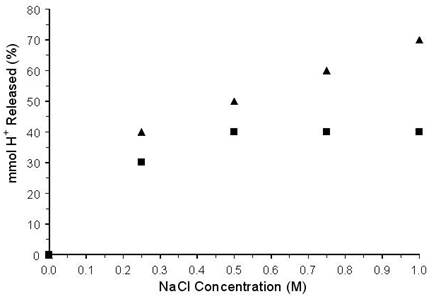
The increase in solution acidity for HZSM-5 in 0.50 M Na+ was not significantly different from the increase observed when higher Na+ concentrations were employed (Figure 2). On the other hand, results for HY indicated that increasing the concentration of Na+ from 0.25 M to 1 M produced an increase in the amount of acidity imparted to the solution. Although not shown in Figure 2, 5 M Na+ resulted in similar solution acidity as was found by using 1 M Na+, suggesting that the amount of H+ exchanged from HY had reached a maximum at 1 M Na+. Although the mmol H+ transferred to the aqueous solution was relatively constant above 0.50 M NaCl for HZSM-5, the corresponding acidities for solutions containing HY increased up to 1 M NaCl. This may be explained by differences in acid concentrations. Fresh HY (2.3 mmol H+/g) had more than twice the H+ contained in the same mass of HZSM-5 (0.9 mmol H+/g). The H+ released from HY and HZSM-5 in 1 M Na+ resulted in 80 and 82% reducing sugar yields for starch hydrolysis, respectively.
3.4. Cellulose Hydrolysis in Ionic Liquid
The dramatic increase in hydrolysis activity observed in the presence of Na+ suggests that ion exchange is an important process when using zeolite catalysts with liquid reaction media. In fact, previous studies have shown that ion exchange rates are heavily dependent on water content [24, 25]. Thus, an ion exchange mediated catalytic activity increase would be expected for reactions in high ionic strength media, such as ionic liquids. To test the influence of ionic liquid media on zeolite acidity, HZSM-5 and HY catalysts were mixed with 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl) and heated at 105 oC for one hour. Titrations of the zeolites after they were exposed to the ionic liquid revealed significant loss of protons from the solids, presumably by an ion exchange mechanism [5]. Fresh HZSM-5 and HY catalysts were found to contain 1.1 and 2.9 mmol H+/g respectively. After exposure to the ionic liquid, these catalysts contained only 0.2 and 1.2 mmol H+/g respectively. The most likely cause for these acidity losses was ion exchange of the imidazolium cation (EMIM+) for H+ [5].
X-ray diffraction measurements of solid state [EMIM]Cl indicated that the cation has dimensions of approximately 3.5 x 3 x 6.5 Å (including hydrogens). Based on these dimensions and the channel openings for HZSM-5 (5.3 x 5.6 Å) and HY (7.4 x 7.4 Å), it is reasonable to expect that EMIM+ could enter the channels of both zeolites. EMIM+ should be able to enter the HY channels at any orientation. The smaller HZSM-5 channel, on the other hand, would restrict entry of EMIM+ to specific orientations.
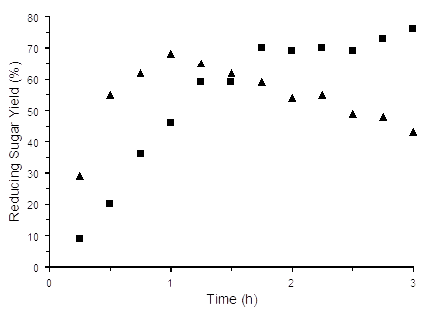
Under reaction conditions similar to those used for aqueous starch hydrolysis, reducing sugar yields from HY catalyzed cellulose hydrolysis increased faster with increasing temperature than HZSM-5 during the first hour (Figure 3). After 1 hour, HY reducing sugar yield decreased, which was likely due to acid catalyzed sugar decomposition. In contrast, HZSM-5 reducing sugar yield increased steadily until about 1.5 hours, and then leveled off. The trends shown in Figure 3 are consistent with more protons being released from HY than from HZSM-5 by ion exchange with EMIM+.
X-ray diffraction (XRD) was used to determine if the HY and HZSM-5 structures were significantly altered by exposure to [EMIM]Cl. Fresh and spent catalysts (e.g. before and after cellulose hydrolysis in [EMIM]Cl, respectively) were analyzed. Comparing the diffraction patterns for the zeolites before and after reactions revealed 95 and 98% similarities for HY and HZSM-5 structures respectively.
HZSM-5 and HY zeolites were also analyzed by DRIFTS prior to and after [EMIM]Cl exposure. Figure 4 shows the resulting infrared spectra and the ionic liquid spectrum for reference. Several overlapping absorbance bands near 3000 cm-1 appear in the [EMIM]Cl spectrum. These spectral features represent stretching vibrations for imidazole alkyl groups.
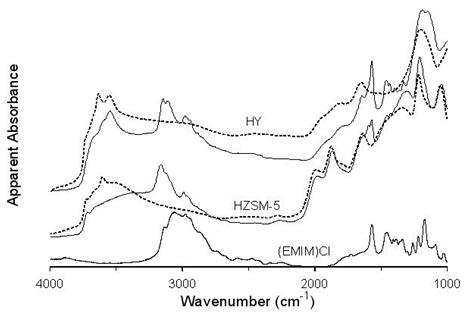
For HY, two characteristic O-H stretching vibration bands at 3650 and 3550 cm-1 are observed that are indicative of Bronsted acidity [26-28]. For HZSM-5, a broad band centered near 3600 cm-1 denotes O-H stretching vibrations associated with acid sites [29].
After exposure to [EMIM]Cl, HY and HZSM-5 DRIFTS spectra exhibited absorbances near 3000 cm-1, which can be attributed to EMIM+. Bands at 2980, 3107, and 3146 cm-1 appear in the HY spectrum. The HZSM-5 spectrum obtained after exposure to [EMIM]Cl contains overlapping bands at 2988, 3123, and 3161 cm-1. Although the EMIM+ band frequencies are somewhat different for HY and HZSM-5, the shapes of the absorbance envelopes in this spectral region were similar for both catalysts. However, both spectra were significantly different from the corresponding envelope in the [EMIM]Cl spectrum. The C-H stretching vibrations for the immadazole ring carbons are most likely responsible for the bands above 3100 cm-1 [30]. The lower wavenumber band in this region can be assigned to the C-H stretching vibration associated with the carbon atom that is attached to both ring nitrogens. The higher wavenumber band is most likely associated with C-H stretching vibrations associated with the other two ring carbons. In [EMIM]Cl, hydrogen bonding between the ring C-H functional groups and Cl- results in a significant shift of the C-H stretching vibrations to lower wavenumber [30]. The higher C-H stretching wavenumbers for the zeolite samples indicate that [EMIM]+ is trapped within the zeolite channels and that Cl- is not available for hydrogen bonding, confirming that the ionic liquid was not simply mixed with the zeolites. In fact, the cation had migrated into the zeolite channels. This conclusion is consistent with a previous report that the average pore diameter for HY decreased after exposure to an ionic liquid [5]. The slight differences in absorption maxima wavenumbers for immadazole ring C-H stretching vibrations for HZSM-5 and HY indicate that the EMIM+ environments within zeolite channels are somewhat different. This is most likely due to differences in zeolite channel dimensions.
4. Conclusions
When used in liquid media, zeolite acid catalysts can operate by two modes. When substrates are small enough to enter framework channels, they can interact directly with solid acid sites. When solutions contain cations that can exchange for zeolite protons, the reaction medium becomes acidified and acid catalyzed reactions can take place outside of zeolite channels. Water reactions with zeolite frameworks can lead to loss of acid sites. This effect is more pronounced for HY than HZSM-5 and the degree of acid site loss can be correlated with Si/Al ratios. Higher Si/Al ratios result in greater hydrophobicity within zeolite channels and less water penetration [21]. Namba et al. [2] investigated the effects of Si/Al ratio on ethyl acetate hydrolysis and found that catalytic activity was optimum for HZSM-5 with a Si/Al ratio of 47. Below this value, acid site destruction due to reactions with water may dominate even though the acid site density increases due to increased framework Al content. Above the optimum Si/Al ratio, a lower acid site density may be responsible for decreased catalytic activity. The low Si/Al ratio for the HY used for studies described here would be expected to result in substantial water mediated acid site destruction. In fact, no acidity was derived from this catalyst for ethyl acetate or starch hydrolysis in aqueous solutions. In contrast, HZSM-5 (Si/Al = 19) exhibited catalytic activity, so it was affected to a lesser extent than HY.
Results described here suggest that ion exchange effects should be considered when evaluating new solid acid catalysts for biomass hydrolysis. Ion exchange negates the advantage of using a solid acid, which is easily removed by filtration, because it results in an increase in solvated protons, in the same manner that could be achieved more easily by using a liquid acid catalyst. Clearly, biomass processing schemes incorporating reusable solid acid hydrolysis catalysts should provide for proton access to solvated starch and cellulose molecules and a means for removing the "fully protonated" solid acid after hydrolysis. This would eliminate the need for neutralizing solution acidity prior to fermentation.
References
[1] Y.B. Huang and F. Yao, "Hydrolysis of Cellulose to Glucose by Solid Acid Catalysts" Green Chemistry, 2013. 15(5): p. 1095-1111.View Article
[2] S. Namba, N. Hosonuma, and T.Yashima "Catalytic Application of Hydrophobic Properties of High-Silica Zeolites. 1. Hydrolysis of Ethyl-Acetate in Aqueous Solution" Journal of Catalysis, 1981. 72(1): p. 16-20.View Article
[3] P.L. Dhepe, M. Ohashi, S. Inagaki, M. Ichikawa, A. Fukuoka "Hydrolysis of Sugars Catalyzed by Water-Tolerant Sulfonated Mesoporous Silicas" Catalysis Letters, 2005. 102(3-4): p. 163-169.View Article
[4] Y. Jiang, X. Li, Q. Cao, X. Mu "Acid Functionalized, Highly Dispersed Carbonaceous Spheres: an Effective Solid Acid for Hydrolysis of Polysaccharides" Journal of Nanoparticle Research, 2011. 13: p. 463-469.View Article
[5] H. Cai, C. Li, A. Wang, G. Xu, T. Zhang "Zeolite-Promoted Hydrolysis of Cellulose in Ionic Liquid, Insight into the Mutual Behavior fo Zeolite, Cellulose, and Ionic Liquid" Applied Catalysis B: Environmental, 2012. 123-124: p. 333-338. View Article
[6] S.R. Benedict, "A Reagent for the Detection of Reducing Sugars" Journal of Biological Chemistry, 1909. 5(5): p. 485-487. View Article
[7] D.R. White and R.L. White, "Isoconversion Effective Activation Energy Profiles by Variable Temperature Diffuse Reflection Infrared Spectroscopy" Soceity for Applied Spectroscopy, 2008. 62: p. 116-120. View Article
[8] N. Horita, M. Yoshimune, Y. Kamiya, T. Okuhara "CS2.5H0.5PW12O40 Bonded to an Amine Functionalized SiO2 as an Excellent Water-Tolerant Solid Acid" Chemistry Letters, 2005. 34(10): p. 1376-1377. View Article
[9] K. Inumaru, T. Ishihara, Y. Kamiya, T. Okuhara, S. Yamanaka "Water-Tolerant, Highly Active Solid Acid Catalysts Composed of the Keggin-Type Polyoxometalate H3PW12O40 Immobilized in Hydrophobic Nanospaces of Organomodified Mesoporous Silica" Angewandte Chemie International Edition, 2007. 46(40): p. 7625-7628. View Article
[10] T. Okuhara, "Water-Tolerant Solid Acid Catalysts" Chemical Reviews, 2002. 102(10): p. 3641-3665. View Article
[11] L.S. Li, Y. Yoshinaga, and T. Okuhara "Unusual acceleration of acid-catalyzed reactions by water in the presence of Mo/Zr mixed oxides calcined at high temperatures" Physical Chemistry Chemical Physics, 2002. 4(24): p. 6129-6136. View Article
[12] A. Tarafdar, A.B. Panda, N.C. Pradhan, P. Pramanik "Synthesis of Spherical Mesostructured Zirconium Phosphate with Acidic Properties" Microporous and Mesoporous Materials, 2006. 95(1-3): p. 360-365. View Article
[13] Y. Kamiya, S. Sakata, Y. Yoshinaga, R. Ohnishi, T. Okuhara "Zirconium Phosphate with a High Surface Area as a Water-Tolerant Solid Acid" Catalysis Letters, 2004. 94(1-2): p. 45-47. View Article
[14] S. Manjare and A. Ghoshal "Comparison of Adsorption of Ethyl Acetate on Activated Carbon and Molecular Sieves 5A and 13X" Journal of Chemical & Engineering Data, 2006. 51: p. 1185-1189.View Article
[15] R.E. Mesmer, W.L. Marshall, D.A. Palmer, J.M. Simonson, H.F. Holmes "Thermodynamics of Aqueous Association and Ionization Reactions at High Temperatures and Pressures" Journal of Solution Chemistry, 1988. 17(8): p. 699-718.View Article
[16] R.S. Drago, S.C. Dias, M. Torrealba, L. de Lima "Calorimetric and Spectroscopic Investigation of the Acidity of HZSM-5" Journal of the American Chemiscal Society, 1997. 119(19): p. 4444-4452. View Article
[17] G.T. Kerr, "Some Comments on Bolton's "Deammination Ammonium Y" Zeolite" Journal of Catalysis, 1974. 35(3): p. 476-477.
[18] L.M. Parker, D.M. Bibby, and G.R. Burns, "Interaction of Water with the Zeolite HY, Studied by FTIR" Zeolites, 1991. 11: p. 293-297. View Article
[19] R.D. Shannon, K.H. Gardner, R.H. Staley, G. Bergeret, P. Gallezot, A. Auroux "The Nature of the Nonframework Aluminum Species formed during the Dehydroxylation of H-Y" Journal of Physical Chemistry, 1985. 89: p. 4778-4788. View Article
[20] J. Scherzer and J.L. Bass, "Infrared Spectra of Ultrastable Zeolites Derived from Type Y Zeolites" Journal of Catalysis, 1973. 28: p. 101-115. View Article
[21] R. Gounder and M.E. Davis, "Beyond Shape Selective Catalysis with Zeolites: Hydrophobic Void Spaces in Zeolites Enable Catalysis in Liquid Water" American Institute of Chemical Engineers (AiChe), 2013. 59(9): p. 3349-3357. View Article
[22] G.S. Nivarthy, K. Seshan, and J.A. Lercher, "The Influence of Acidity on Zeolite H-BEA Catalyzed Isobutane/n-Butene Alkylation" Microporous and Mesoporous Materials, 1998. 22(1-3): p. 379-388. View Article
[23] B. Silva, H. Figueiredo, O.S.G.P. Soares, M.F.R Pereira, J.L. Figueiredo, A.E. Lewandowska, M.A. Bañares, I.C. Neves, T. Tavares "Evaluation of Ion Exchange-Modified Y and ZSM-5 Zeolites in Cr(VI) Biosorption and Catalytic Oxidation of Ethyl Acetate" Applied Catalysis B: Environmental, 2012. 117-118: p. 406-413. View Article
[24] A. Dyer and R.B. Gettins, "Mobility of Cations in Synthetic Zeolites with Faujasite Framework 3. Self-Diffusion of Cations into X-Zeolites and Y-Zeolites from Non-Aqueous Solutions" Journal of Inorganic and Nuclear Chemistry, 1970. 32: p. 2401-2410. View Article
[25] H. Gaus and W. Lotze, "Kinetics of Sr-Ba and Sr-Ca Ion-Exchange in Synthetic Zeolite A" Journal of Physical Chemistry, 1981. 85: p. 79-84. View Article
[26] R.A.v. Santen, "Theory of Bronsted acidity in zeolites" Studies in Surface Science and Catalysis, 1994. 85: p. 273-294. View Article
[27] J.C. Lavalley, R. Anquetil, J. Czyzniewska, M. Ziolek "Use of Pyridine as a Probe for the Determination, by IR spectroscopy, of the Bronsted Acid Strength of M(I)HNaY Zeolites" Journal of the Chemical Society, Faraday Transactions, 1996. 92(7): p. 1263-1266. View Article
[28] P. Jacobs and J. Uytterhoeven, "Assignment of the Hydroxyl Bands in the Infrared Spectra of Zeolites X and Y" Journal of the Chemical Society, Faraday Transactions, 1972. 69: p. 359-372. View Article
[29] P.A. Jacobs and R. Vonballmoos, "Framework Hydroxyl Groups of H-ZSM-5 Zeolites" Journal of Physical Chemistry, 1982. 86(15): p. 3050-3052. View Article
[30] C.J. Dymek and J.J.P. Stewart, "Calculation of Hydrogen-Bonding Interactions between Ions in Room-Temperature Molten Salts" Inorganic Chemistry, 1989. 28: p. 1472-1476. View Article